
A biocompatible nanoparticle-based approach to inhibiting renal ischemia reperfusion injury in mice by blocking thrombospondin-1 activity
Yue Hou1,2,3, Shuhan Liu1,2,3, Xiandi Meng1,2,3, Yanbao Xin1,2,3, Jialiang Wang1,2,3, Tianmeng Sun1,2,3,4, Yong-Guang Yang1,2,3,4.
1Key Laboratory of Organ Regeneration and Transplantation of the Ministry of Education, Jilin University, Changchun, People's Republic of China; 2The First Hospital, Jilin University, Changchun, People's Republic of China; 3National-local Joint Engineering Laboratory of Animal Models for Human Diseases, Changchun, People's Republic of China; 4International Center of Future Science, Jilin University, Changchun, People's Republic of China
Background: Ischemia-reperfusion injury (IRI) impairs graft function and promotes acute rejection following renal allotransplantation. Thrombospondin-1 (TSP1) is a matricellular protein produced by a variety of cells, and its expression is upregulated in tissues following IRI. CD47 is a ubiquitously expressed transmembrane protein that serves as a ligand for SIRPα and is a signaling receptor of TSP1. Ligation of CD47 by TSP1 has been shown to play an important role in mediating renal IRI. TSP1 also activates transforming growth factor beta (TGF-β), another factor contributing to IRI. In this study, we sought to develop a biocompatible nanoparticle-based approach for inhibiting renal IRI by blocking TSP1 activity.
Methods: PLGA nanoparticle (NP) preparation and conjugation of TSP1-binding peptides (LSKL, a sequence of TGF-β) to NPs are elucidated in Fig. 1. The ability of LSKL-conjugated NPs (LSKL/NPs) to bind TSP1 was tested in vitro using fluorescence AF647-conjugated TSP1. The potential of LSKL/NPs to protect against IRI was evaluated in BALB/c mice, in which IRI was induced by clamping the left renal pedicle for 28 min following surgical removal of the right kidney. PBS, free LSKL and NPs (without LSKL) were used as controls in these experiments.
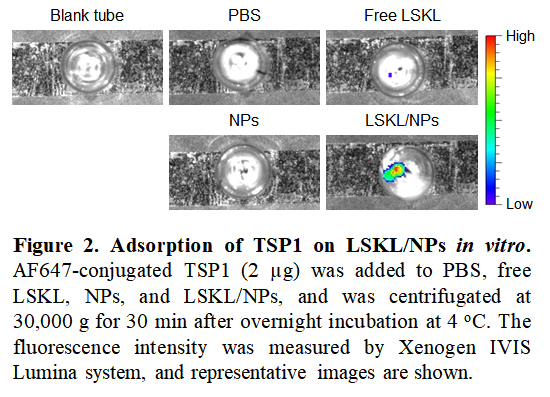
Results: We first determined the ability of LSKL/NPs to bind/adsorb TSP1 by measuring fluorescence intensity of NPs after overnight incubation with AF647-TSP1. Imaging analysis revealed strong fluorescence signals in LSKL/NPs, while only limited/background signals were detected in free LSKL and NPs compared to PBS controls (Fig. 2), indicating that LSKL conjugation confers the ability of NPs to bind/adsorb TSP1.
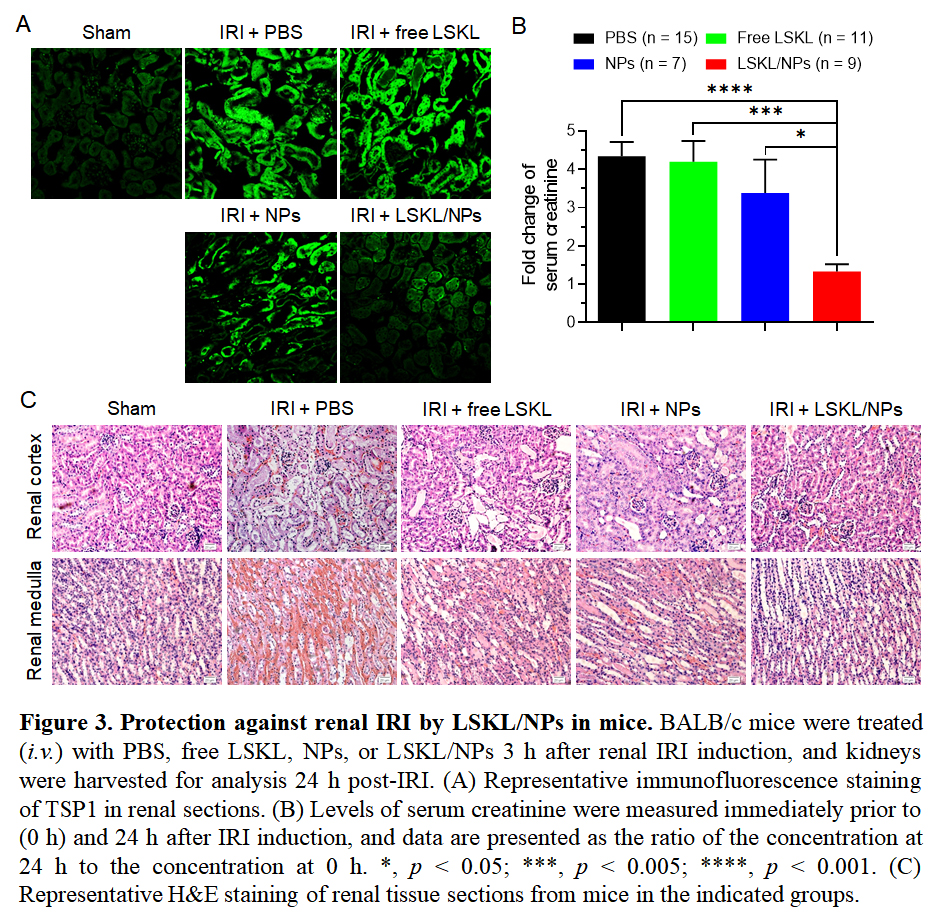
Because TSP1 adsorption to NPs may result in TSP1 clearance and loss of function due to conformational changes, treatment with LSKL/NPs is expected to inhibit TSP1 activity and hence IRI. To address this question, we compared renal IRI in mice that were treated with PBS, free LSKL, NPs, or LSKL/NPs 3 h after IRI induction. Immunofluorescence staining of tissue sections showed that IRI induced a significant and rapid increase in TSP1 production in the kidney, which was markedly inhibited in mice treated with LSKL/NPs (Fig. 3A). Furthermore, treatment with LSKL/NPs almost completely inhibited IRI-induced elevation of serum creatinine, while serum creatinine levels in mice treated with fee LSKL or NPs were comparable to those of PBS controls (Fig. 3B). In line with this observation, severe tubular destruction and interstitial hemorrhage, which were clearly detected in kidneys from mice treated with PBS, free LSKL or NPs, were markedly ameliorated in LSKL/NP-treated mice (Fig. 3C).
Conclusion: Our data demonstrate that LSKL/NPs provide an efficient means of adsorbing/neutralizing TSP1, hence inhibiting renal IRI. While other mechanisms may also be involved, protection against IRI by LSKL/NPs is assumed to be largely conferred by its ability to block TSP1-induced CD47 and TGF-β activation.